GLIOBLASTOMA MULTIFORME
 Glioblastoma multiforme (GBM) is by far the most common and most
malignant of the glial tumors.
The graded anaplasia of the glioma group reaches its extreme in
glioblastoma multiforme. Gliomas comprise a heterogeneous group
of neoplasms that differ in location within the central nervous
system, in age and sex distribution, in growth potential, in
extent of invasiveness, in morphological features, in tendency
for progression, and in response to treatments.
Glioblastoma multiforme (GBM) is by far the most common and most
malignant of the glial tumors.
The graded anaplasia of the glioma group reaches its extreme in
glioblastoma multiforme. Gliomas comprise a heterogeneous group
of neoplasms that differ in location within the central nervous
system, in age and sex distribution, in growth potential, in
extent of invasiveness, in morphological features, in tendency
for progression, and in response to treatments.
Most authors consider this lesion to be
a neoplasm of astrocytes because (1) the glioblastoma merges as
a clinical entity with the two better-differentiated
astrocytomas and anaplastic astrocytomas, (2) pathologically the
glioblastoma sometimes evolves out of a better-differentiated
astrocytic tumor, and (3) it often contains neoplastic
astrocytes. It is recognized, however, that many glioblastomas
appear to arise de novo, some are totally undifferentiated, and
a rare glioblastoma evolves out of another glioma such as an
oligodendroglioma. Glioblastomas can be classified as primary or
secondary. Primary glioblastoma multiforme accounts for the vast
majority of cases (60%) in adults older than 50 years. These
tumors manifest de novo (i.e., without clinical or
histopathologic evidence of a preexisting, less-malignant
precursor lesion), presenting after a short clinical history,
usually less than 3 months.
Secondary glioblastoma multiforme (40%) typically develop in
younger patients (< 45 y) through malignant progression from a
low-grade astrocytoma (WHO grade II) or anaplastic astrocytoma
(WHO grade III). The time required for this progression varies
considerably, ranging from less than 1 year to more than 10
years, with a mean interval of 4-5 years. Increasing evidence
indicates that primary and secondary glioblastomas constitute
distinct disease entities that evolve through different genetic
pathways, affect patients at different ages, and differ in
response to some of the present therapies. Of all the astrocytic
neoplasms, glioblastomas contain the greatest number of genetic
changes, which, in most cases, result from the accumulation of
multiple mutations.
Over the past decade, the concept of different genetic pathways
leading to the common phenotypic endpoint (ie, GBM) has gained
general acceptance. Genetically, primary and secondary
glioblastomas show little overlap and constitute different
disease entities. Studies are beginning to assess the prognoses
associated with different mutations. Some of the more common
genetic abnormalities are described as follows:
Loss of heterozygosity (LOH): LOH on chromosome
arm 10q is the most frequent gene alteration for both primary
and secondary glioblastomas; it occurs in 60-90% of cases. This
mutation appears to be specific for glioblastoma multiforme and
is found rarely in other tumor grades. This mutation is
associated with poor survival. LOH at 10q plus 1 or 2 of the
additional gene mutations appear to be frequent alterations and
are most likely major players in the development of
glioblastomas.
p53: Mutations in p53, a tumor suppressor gene, were among the
first genetic alterations identified in astrocytic brain tumors.
The p53 gene appears to be deleted or altered in approximately
25-40% of all glioblastoma multiforme, more commonly in
secondary glioblastoma multiforme. The p53 immunoreactivity also
appears to be associated with tumors that arise in younger
patients.
Epidermal growth factor receptor (EGFR) gene: The EGFR gene is
involved in the control of cell proliferation. Multiple genetic
mutations are apparent, including both overexpression of the
receptor as well as rearrangements that result in truncated
isoforms. However, all the clinically relevant mutations appear
to contain the same phenotype leading to increased activity.
These tumors typically show a simultaneous loss of chromosome 10
but rarely a concurrent p53 mutation. Overexpression or
activation mutations in this gene are more common in primary
glioblastoma, with mutations appearing in 40-50% of these
tumors. One such common variant, EGFRvIII, has shown promise as
a target for kinase inhibitors, immunotoxins, and peptide
vaccines.
MDM2: Amplification or overexpression of MDM2 constitutes an
alternative mechanism to escape from p53 -regulated control of
cell growth by binding to p53 and blunting its activity.
Overexpression of MDM2 is the second most common gene mutation
in glioblastoma multiforme and is observed in 10-15% of
patients. Some studies show that this mutation has been
associated with a poor prognosis.
Platelet-derived growth factor–alpha (PDGF-alpha) gene: The PDGF
gene acts as a major mitogen for glial cells by binding to the
PDGF receptor (PDGFR). Amplification or overexpression of PDGFR
is typical (60%) in the pathway leading to secondary
glioblastomas.
PTEN: PTEN (also known as MMAC and TEP1) encodes a tyrosine
phosphatase located at band 10q23.3. The function of PTEN as a
cellular phosphatase, turning off signaling pathways, is
consistent with possible tumor-suppression action. When
phosphatase activity is lost because of genetic mutation,
signaling pathways can become activated constitutively,
resulting in aberrant proliferation. PTEN mutations have been
found in as many as 30% of glioblastomas, more commonly in
primary glioblastoma multiforme.
Less frequent but more malignant mutations include the
following:
MMAC1-E1 - A gene involved in the progression of gliomas to
their most malignant form
MAGE-E1 - A glioma-specific member of the MAGE family that is
expressed at up to 15-fold higher levels in glioblastoma
multiforme than in normal astrocytes
NRP/B - A nuclear-restricted protein/brain, which is expressed
in neurons but not in astrocytes (NRP/B mutants are found in
glioblastoma cells.)
Additional genetic alterations in primary glioblastomas include
p16 deletions (30-40%), p16INK4A, and retinoblastoma (RB) gene
protein alterations. Progression of secondary glioblastomas
often includes LOH at chromosome arm 19q (50%), RB protein
alterations (25%), PTEN mutations (5%),
deleted-in-colorectal-carcinoma gene (DCC) gene loss of
expression (50%), and LOH at 10q.
Frequency
Glioblastoma multiforme is the most frequent primary brain
tumor, accounting for approximately 12-15% of all intracranial
neoplasms and 50-60% of all astrocytic tumors. In most European
and North American countries, incidence is approximately 2-3 new
cases per 100,000 people per year.
In any site, the glioblastoma expresses its outright
anaplasia as the induration and sometimes fleshy grayness of
high cellularity and the hemorrhagic necrosis of rapid
growth. These features are most apparent in the cerebral
hemispheres of adults, where the characteristically
deep-seated lesion is most common, but can also be observed
in the brain stem or in the rare lesion of the spinal cord.
The gray fleshiness is typical of any markedly cellular
lesion and is often most apparent in areas of invaded
cortical gray matter. The necrotic regions are usually more
centrally placed and are known for distinctive variegation
by reds, browns, and yellows. Unlike the well-differentiated
astrocytoma, the mass often appears well defined, and any
cysts are filled with dirty brown, rather than clear yellow,
fluid. The neoplastic cells diffuse freely through the white
matter and can funnel into fiber pathways such as the corpus
callosum, internal capsule, or anterior commissure.
Expansion into the opposite cerebral hemisphere then
produces the classic butterfly lesion.
At surgery, the circumscription of the glioblastoma can be
sufficiently pronounced, especially in the giant cell
variant. to suggest a metastatic carcinoma. The deposition
of collagen, either as a reactive process or as part of a
neoplastic proliferation of fibroblasts ("gliosarcoma"),
can enhance this definition and produce a discrete, firm
mass. In general, however, the glioblastoma's infiltrating
border, large size, and prominent central area of necrosis
are distinguishing features. The soft necrotic character of
some glioblastomas can be simulated by the primary cerebral
lymphoma or the occasional infarct that comes to surgery.
The latter shows prominent softening of the cerebral cortex
and lacks the fleshy grayness of the glioma. A rare patient
with a glioblastoma has the acute effects of a large intratumoral hemorrhage, although the incidence of this
event in this neoplasm is much lower than in metastatic
neoplasms such as malignant melanoma and choriocarcinoma.
The incidence is high enough, however, to justify pathologic
study of tissue fragments adherent to intracranial
hematomas.
Microscopically, most of the viable
neoplastic cells in the glioblastoma are concentrated in a
region of high cellularity that circumscribes a central
area of necrosis. In less-advanced neoplasms, only scattered
smaller areas of necrosis may be seen. The cellular areas
are apparent macroscopically as the fleshy rim and radiographically as the ring of contrast enhancement.
Peripherally, the neoplastic cells diffuse away into the
surrounding edematous brain for distances that must be
considerable in light of the failure of large en bloc
resections to cure the lesion. In contrast to the
well-differentiated astrocytoma, calcification is rare.
The cytologic composition of the glioblastoma
includes a remarkably heterogeneous array of cell types
such as fibrillary astrocytes, gemistocytes, larger and
more pleomorphic astrocytes, and large bizarre cells with
extreme pleomorphism. For the most part these
elements have characteristics generally attributed to
astrocytes; that is, they have stellate processes as in the
fibrillary and gemistocytic astrocytes and a prominent
glassy cytoplasm as in the more bizarre astrocytes and giant
cells. With immunoperoxidase staining, the fibrillary and gemistocytic astrocytes are often positive, whereas the pink
cytoplasm of the other cells shows a variable positivity. By
electron microscopy, the positivity with immunoperoxidase is
correlated with the presence of the cytoplasmic "glial"
filaments. These are numerous in the fibrillary astrocytes
and unpredictably present in the large glassy cytoplasm of
the remaining cell types.
A cell that often predominates
in the glioblastoma but is present also in limited numbers
in the anaplastic astrocytoma is a small anaplastic form
with a round to elongated nucleus. These cells proliferate
to a remarkably high density and are usually responsible
for gray, fleshy areas. They also are extremely mobile and
diffuse freely through the corpus callosum or other fiber
tracts, invade the cortex to surround neurons, and aggregate
in a subpial position. In addition, they often are prominent
about areas of necrosis, suggesting that this distinctive
peripheral concentration of cells could be a consequence
of their motility by which they accumulate
at the edge because they are unable to pass through the
necrotic center.
Some glioblastomas contain
many large, bizarre cells, and such neoplasms have been
variously referred to as giant cell glioblastoma,
giant cell fibrosarcoma, or a type of gliosarcoma. Most
authors would classify most of these lesions as gliomas and
have noted that, in spite of their alarming microscopic
appearance, the survival rate for patients with these
lesions is somewhat more favorable than for those with the
typical glioblastoma. This may relate to
their well-circumscribed nature and ease of surgical
excision and/or to the reduced biological aggressiveness of
bizarre giant cells.
Reactive fibroblasts populate
some glioblastomas, whereas neoplastic fibroblasts
proliferate in others. In either setting, mesenchymal
cells are distinguished from the glial component by the
former's polarity, association with reticulin and collagen,
and absence of GFAP. A higher incidence in the
temporal lobes has been suggested for the mixed neoplastic
lesions known as gliosarcomas. It is worth noting that the
distinction between reactive and neoplastic fibroblasts is
often quite subjective: what is a gliosarcoma to one
observer may be a glioblastoma with reactive fibrosis to
another. The distinction appears largely academic, however,
because the presence of a sarcomatous component does not
seem to modify the prognosis.
For diagnostic purposes, the histologic diagnosis of
glioblastoma is usually made as much on the basis of two
distinctive secondary features as on cytologic
characteristics. The first of these is
vascular proliferation, by which vascular cells divide to
produce coiled masses resembling renal glomeruli. These new vessels often have a directional
orientation, as the coils point toward a common site
such as an area of high cellularity or necrosis. A response
to an angiogenic factor liberated by the neoplasm
has been suggested. The phenomenon is often referred to as
endothelial
proliferation or
vascular
endothelial proliferation (VEP); however, several
immunohistochemical studies
have demonstrated that other vascular elements, such as
vascular smooth muscle cells, pericytes, and perivascular
fibroblasts, are major participants in the proliferative
process. The term
microvascular proliferation, which has achieved some
currency, is more accurate.
Microvascular proliferation is an important
feature differentiating glioblastoma multiforme from the welldifferentiated fibrillary astrocytoma and the
anaplastic astrocytoma, although a small amount of this
proliferation is acceptable within the latter lesion.
Although characteristic of the glioblastoma, microvascular
proliferation is found also in limited extent in other
neoplasms, such as pilocytic astrocytoma, cerebellar
astrocytoma, oligodendroglioma, and medulloblastoma.
The second diagnostically helpful feature in glioblastoma is
necrosis. This feature,
with or without associated pseudopalisading of neoplastic
cells, is a firm differential point distinguishing
glioblastoma from anaplastic astrocytoma. In lesions that
have been previously treated with radiation therapy, of
course, necrosis may not have this diagnostic value. A
distinctive feature of necrotic areas in glioblastoma
multiforme, especially the smaller foci, is a concentration
of neoplastic cells that jostle with one another at the
periphery. Because these cells are often elongated and
oriented perpendicularly to their tangents with the
necrotic area, the term
palisade
or pseudopalisade
is applied. In a cerebral hemispheric glioma, this
pseudopalisading is virtually diagnostic of glioblastoma.
Neither the presence nor the
absence of pseudopalisading, however, affects the prognostic
or diagnostic value of necrosis.
The pathologic diagnosis of the glioblastoma is usually not
difficult in generous specimens but can be problematic in
small ones. The pathologist's need for an adequate specimen
cannot be overemphasized. More diagnostic problems can be
resolved by larger specimens than by application of special
stains, including available immunologic techniques. Although
purists contend that glioblastoma should be diagnosed only
in the presence of necrosis and/or vascular proliferation,
in practice there is a justifiable temptation in surgical
material to diagnose glioblastoma multiforme on the basis of
extreme cellularity or pleomorphism alone, especially in the
face of typical clinical and radiographic findings. This is
not condoned in the less cellular lesions, however. The size
of some small specimens also makes it difficult to exclude
other malignant neoplasms such as a metastatic carcinoma.
For this reason it is desirable to submit to the pathologist
specimens from the edge of the most cellular areas. Such
tissues define the relationship of the neoplasm to the
surrounding brain-a relationship that in the case of glioblastoma is one of
diffuse infiltration and in metastatic carcinoma
is the expansion of a cohesive mass. The diagnostically
helpful microvascular proliferation and necrosis with pseudopalisading are also often prominent in this
peripheral region. A positive GFAP stain indicates a glial,
rather than an epithelial, neoplasm, but, like other
special stains, it is often negative in the anaplastic
lesion where its diagnostic value is most needed.
Radio- and chemotherapy produce marked changes in the
glioblastoma that may be encountered at reoperation.
Radiotherapy destroys small cells, leaving better
differentiated astrocytes behind, and induces pleomorphism
in residual neoplastic or reactive glia. Macroscopically,
such treated lesions in remission are discrete, fibrotic,
necrotic, and sometimes calcified. Cysts some-times form.
The subsequent
phenomenon of recurrence relates largely to the regrowth of
the small anaplastic cells discussed above. At this point,
such cells are widely invasive and frequently extend down,
or in the case of brain stem lesions up, the cerebral
peduncles. Perhaps 10 percent of the lesions seed the
ventricular and subarachnoid spaces. In such cases,
cytologic study of the cerebrospinal fluid (CSF) can be an
effective diagnostic tool.
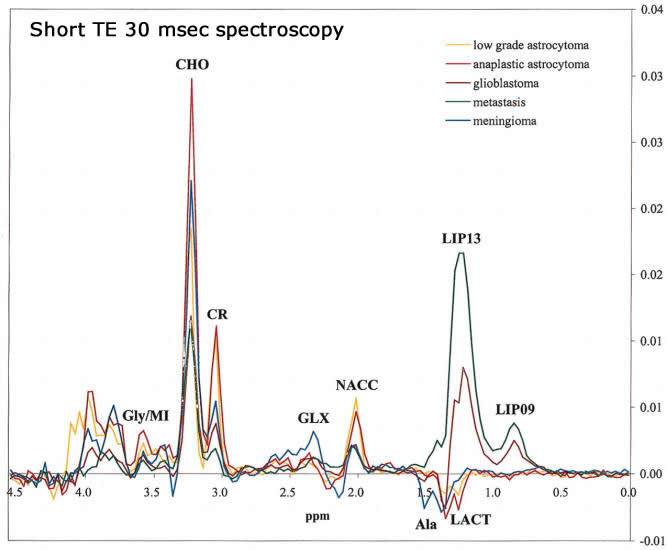
 MRS in Glioblastoma multiforme:
MRS in Glioblastoma multiforme: